Hi, I'm Wes.
I'm a statistical genetics scientist
at Vesalius Therapeutics.
Previously, I trained as a postdoctoral scholar with Xin He and Matthew Stephens in the department of Human Genetics at the University of Chicago. I received my PhD in Bioinformatics and Computational Biology from UNC Chapel Hill. My experience is in statistical genetics, and my research includes developing statistical methods to prioritize causal genes associated with human diseases and other biomedically-relevant traits. I have expertise in causal inference, Bayesian nonparametric models, Bayesian model selection, and generalized linear models, with implementation in the R and Stan statistical computing languages.
I am interested in using cutting-edge statistical methods to positively impact human health.
Research
You can see my full list of publications on Google Scholar. Here are some of the projects I've worked on recently:
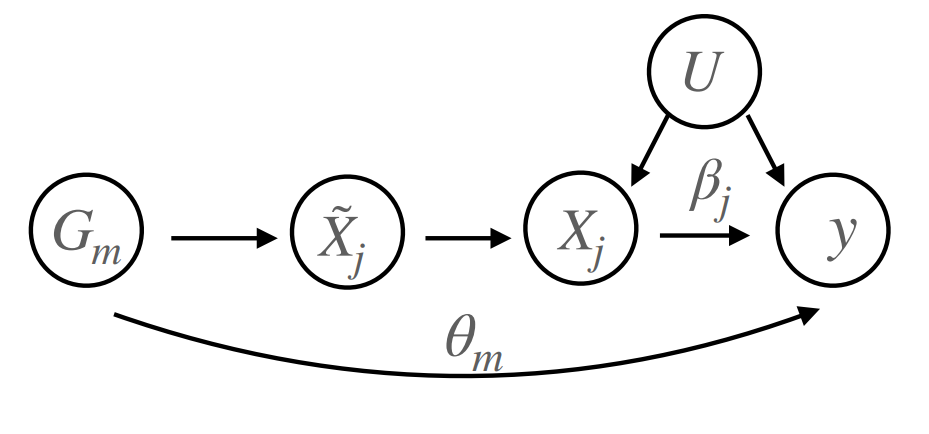
cTWAS
causal-TWAS (cTWAS) is an integrative method for identifying causal genes at GWAS loci using eQTL data. Transcriptome-wide association studies (TWAS) use eQTL to infer gene expression in a GWAS cohort and test (inferred) gene expression for association with a trait. TWAS is useful for nominating candidate genes but prone to false positives due to genetic confounding. Genetic confounding occurs when non-causal genes are linked with causal variants or genes that affect the trait through another mechanism. Motivated by causal inference, cTWAS conditions on genetic confounders, jointly modeling sparse effects of all nearby genes and variants in an extended fine-mapping framework. cTWAS substantially reduces false positives relative to alternative methods, making it a reliable tool for linking variants to genes. The manuscript for cTWAS was published in the January 2024 issue of Nature Genetics.
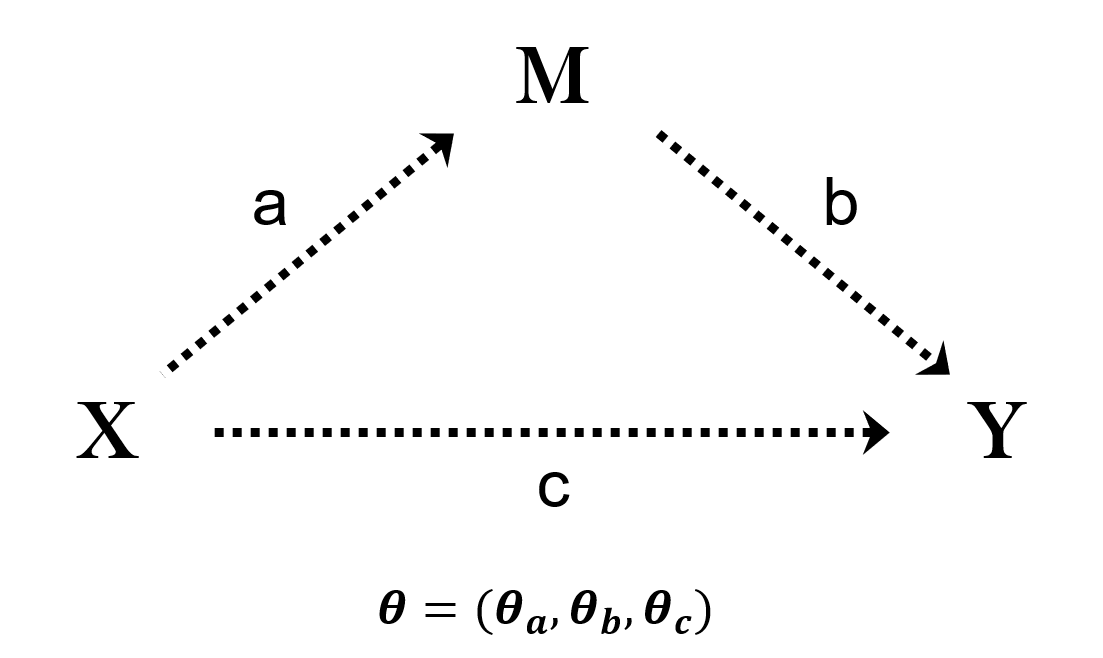
bmediatR
bmediatR is a Bayesian model selection approach for mediation analysis. It explores different causal relationships between a dependent variable (Y), an independent variable (X), and a potential mediator variable (M). This approach distinguishes between partial and complete mediation, and it accommodates grouped predictors in X. Grouped predictors are useful for haplotype-based analyses and other cases when the independent variable is multidimensional, such as modeling multiple variants or non-additive effects. The manuscript for bmediatR was published in the May 2022 issue of PLOS Genetics.
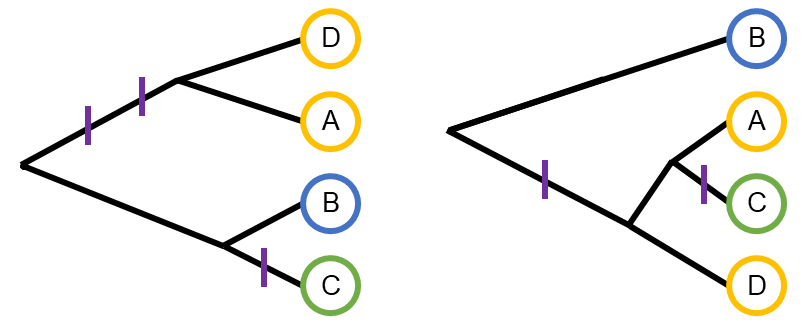
TIMBR
TIMBR is a Bayesian nonparametric model for haplotype-based genetic association. It partitions haplotypes into a potentially smaller number of functional alleles with shared trait effects. This improves haplotype effect estimation and provides useful information about the number of causal variants at a quantitative trait locus. TIMBR partitions haplotypes using a Chinese restaurant process (CRP) and, by leveraging its relationship to the coalescent, generalizes the CRP to allow for tree-structured haplotype relatedness. The manuscript for TIMBR was highlighted in the December 2020 issue of Genetics.
Curriculum Vitae

Here's my CV.
I'm happy to provide additional details about my experience. References are available upon request.